CMU 15-418/618 (Spring 2013) Final Project:
Molecular Dynamics Simulations on a GPU
Original Project Proposal
Checkpoint Report
Schedule and Work Log
Project Summary
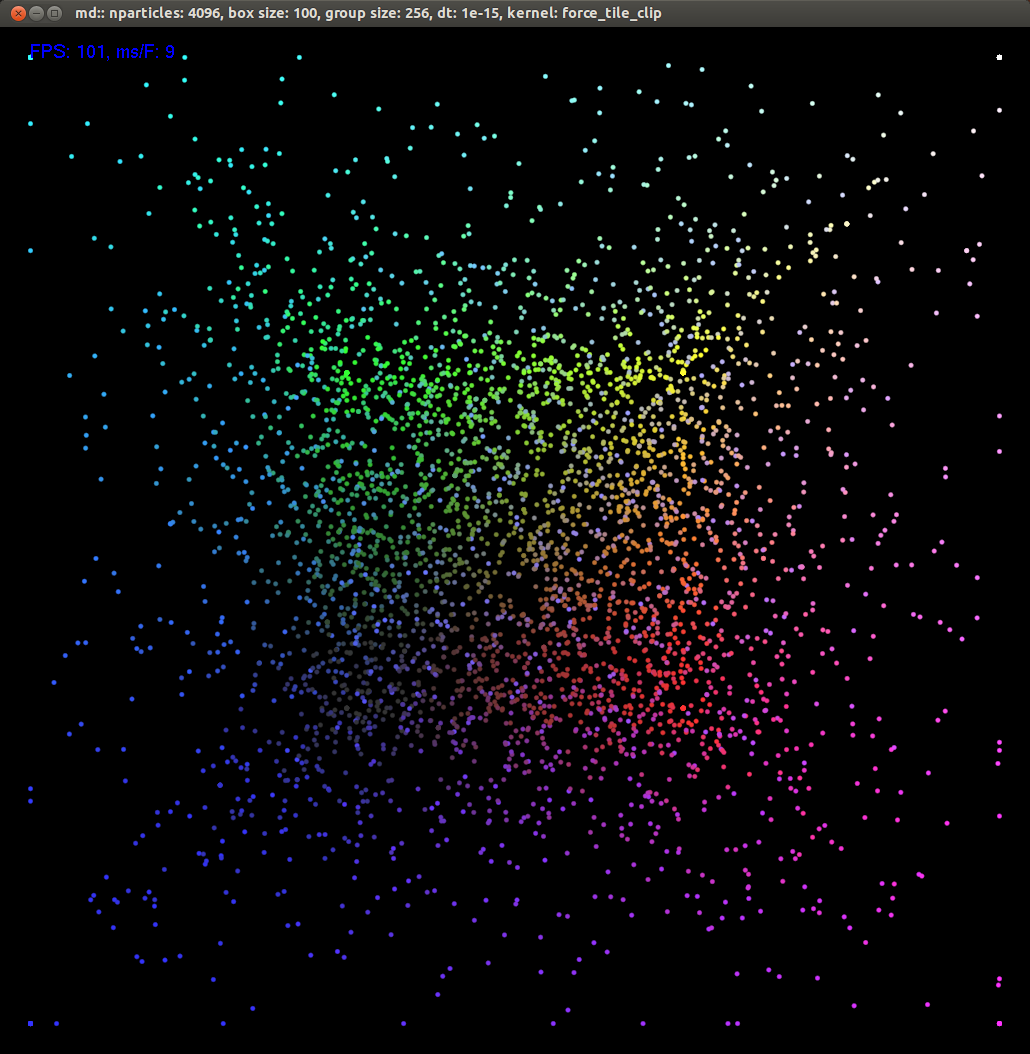
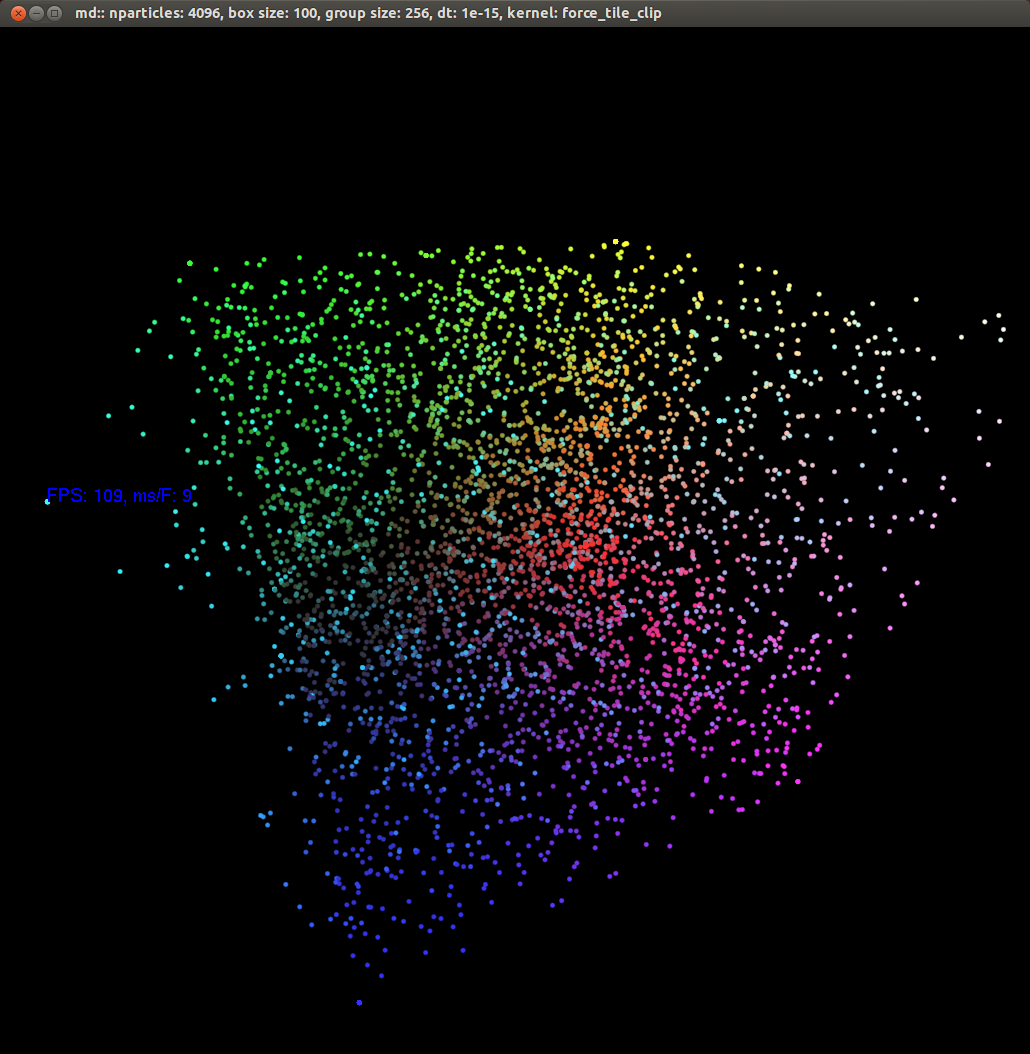
The goal of this project was do develop an effective molecular dynamics simulation in OpenCL to be run on a GPU. Specifically, the focus was on strategies to divide the work of computing the pairwise force interaction between particles. For molecular dynamics, the primary interaction of interest is the Van Der Waals interaction between atoms. I chose to use the relatively simple Lennard Jones potential to model this interaction. The simulation is rendered in OpenGL to visualize the simulation. In the end, I chose to keep each individual simulation very simple and short so that I could analyze its performance over a wide range of parameters. A demonstration of this project would take the form of a video capture instead of a live demo, since I likely won't have a chance to get everything up and running on my laptop (which has inferior power anyway). Since I ran simulations under a wide variety of scenarios, my discussion will be focused on those results.
Background
The problem being solved here is fundamentally identical to an n-body simulation--the only difference is the force of interest. The simulation has two steps: first update forces and then update positions, repeat forever. Updating positions is trivial, since you just need to move each particle based on its current force value. Updating the forces is the interesting part.
In both cases, all $n$ objects interact separately will all $n-1$ other objects, meaning that $O(n^2)$ distinct calculations are required. Therefore, the topic of interest is whether they can be broken up in some manner to allow them to be done faster. Similarly, a common simplification of the problem is to ignore interactions beyond a certain distance, meaning that there are several possibilities to explore.
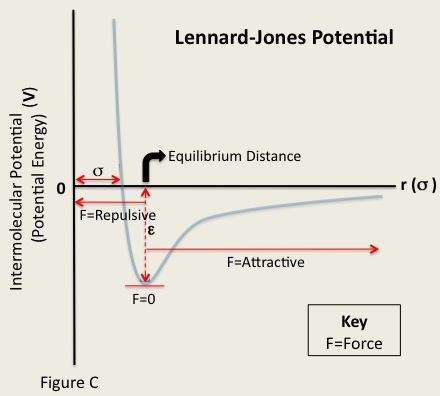
As far as the physics of the problem goes, I'm making a number of simplifying assumptions so that I can focus on the computer science aspect of the problem. One is that all particles are identical (specifically, they're all Xenon atoms). The Lennard Jones potential has two parameters that depend on the particle. The depth of the potential well, equivalently the strength of the interaction, is given by $\epsilon$. The distance that is energetically most favorable is based on $\sigma$. For Xenon, these are $\epsilon = 1.77$ kJ/mol and $\sigma = 4.10$ Angstroms. I don't think that adding support for a heterogeneous group of atoms would add much from the computer science perspective. The force calculation is $$F_{lj}(r) = 24 \epsilon \left[2\frac{\sigma^{12}}{r^{13}} - \frac{\sigma^{6}}{r^{7}}\right]$$ In other words, attractive beyond a certain distance and repulsive within it. This is illustrated in the image below.
Finally, I chose OpenCL for this project because it seems like most of the work done in this area in the past has been to implement parallelism with things like MPI as opposed to GPGPU and SIMD. This seems like a reasonable approach, since the there aren't any data dependencies or critical regions within the force update itself. There's also no communication step to worry about. However, in the context of ignoring long range interactions, divergence may become an issue.
Approach
The naive solution is to consider each particle separately and loop over all other particles to produce the combined force and store the result. Sadly, this doesn't even allow you to take advantage of the symmetry of the problem in any sensible manner. I spent some time on this issue, but the lack of support atomic operations on floats (or float4s) left me with no sensible solution. This approach will be referred to as parallelism over particles.
An alternate approach is parallelism over forces. Although the same amount of work is done, the fundamental difference with this approach is that we can take advantage of memory locality to compute the forces in square tiles (size $p \times p$). Since these tiles all need the same particle positions, they can be loaded into shared memory (the __local address space, in OpenCL terms). So, a further topic of interest brought up in this approach is how large the tile should be. In OpenCL terms, this would be the size of the local group (equivalent to the block size in CUDA terms). A demonstration of how this is broken down is shown below.

Image credit to NVIDIA.
As mentioned previously, another method to improve the speed of the computation (at the expense of accuracy though) is to ignore long range interactions by making them all 0. With all of these possibilities in mind, I wrote four different kernel functions to update the forces.
force_naive: Consider each particle separately and loop over all others.force_naive_clip: Similar to above, but ignore things more than 10 angstroms away.force_tile: Implement the tiling algorithm.force_tile_clip: Like the previous one, but ignores anything more than 10 angstroms away.
Originally, I had planned on using a third strategy of spatial decomposition, where particles are binned and only interactions with adjacent bins are computed. This would improve on the above methods by not even considering particles that are far away (in the above kernels, you still need to look at everyone else, even if you proceed to ignore them). I ended up abandoning this strategy because I couldn't find a suitable way to implement this using fixed-size buffers (the paper I read that presented this in the context of MPI used linked lists, which clearly wouldn't work).
Another strategy that I used to improve the performance of my code was to use OpenCL-OpenGL interoperability to handle the data needed by both OpenCL and OpenGL by storing it in an OpenGL vertex buffer object (VBO) that is shared with OpenCL. This is extremely advantageous because after the data is initialized and sent to the GPU, the memory is never transferred back to the CPU, avoiding a potentially huge bottleneck. This was implemented from the very beginning, so I have no measure of the difference in performance.
Results
I tested the four kernels listed above with a wide variety of parameters to examine their performance. All of these tests were run on my desktop computer (even though we got OpenCL working on the Gates computers, my code was still running into other issues with them). Specs: EVGA GTX 560 Ti with 1 GB DDR5 RAM (specs specs2); AMD Phenom X4 9950 Black Edition (specs specs2); 8 GB DDR2 RAM; Ubuntu 13.04 with the nvidia 304 driver.
Before getting into details, here's an example of what a simulation looks like. The framerate is shown in the top left corner. Color is used primarily to judge depth. Since the camera can be rotated, color is varied in all dimensions. Nothing terribly exciting happens, just a simple demonstration.
Performance was measured based on the time (in ms) required to run
kernel updates to force/position, averaged over 1 second
intervals. The kernel calls and re-rendering the image were handled as
two separate tasks, based on
OpenGL's glutTimerFunc. Since the rendering is just a
nice tool for us, we want to focus as much time as possible on running
the kernel. In other words, the kernel is allowed to execute multiple
times between updating the display. There is a minimum delay of 1ms
between kernel calls and a minimum delay of 50ms between renders. So,
just to be clear, I've used 'frame' to refer to a kernel update, not a
render to the display (I should probably find a better word). It seems
that times tend to cluster around certain data points, but they vary
enough with all inputs to see some key information.
I focused of varying two of the parameters. Seven different values for the number of particles are used, ranging between 1024 and 16384. Seven different values are used for the size of a work group (a.k.a. size of a tile, where applicable), ranging between 16 and 1024. Some other important parameters are: bbox, the particles are constrained to $\pm bbox$ in all three dimensions and was set at 100 angstroms (with one exception); dt, the timestep was set to $10^{-15}$s (1 fs), which is reasonable for this type of simulation. The full data table can be found here. One way that I analyzed these was by plotting the results, grouping all with the same number of particles. One example is shown below, the rest can be found here. Each plot generally reflects the same trends, with some disagreement.
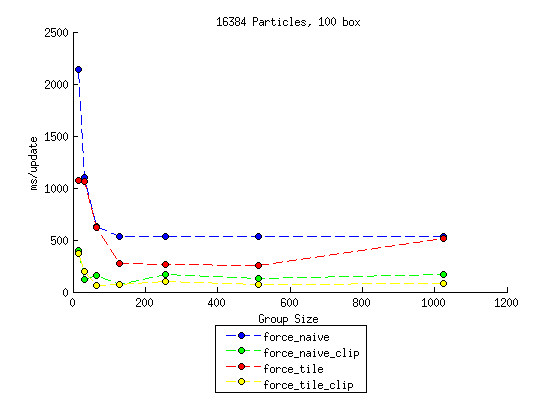
To me, the most striking result here is that ignoring far away particles makes a much greater difference than tiling does. My assumption was that the inherent divergence of this approach would drastically limit its effectiveness. However, the effects of divergence are still observed, given that performance for these generally peaks somewhere in the middle and goes down for the larger group sizes, as one might expect. Overall, the tiled approach generally shows a marginal advantage over the naive approach (when viewed holistically, as individual points vary considerably), but nothing to get too excited about. In the naive approach, there still is a fair amount of locality that can aid the calculation, even if it isn't explicitly designed around this.
Unfortunately, the results are very unclear with respect to what the best group size is. 64, 128, and 256 generally perform well, but there are exceptions. Since the natural SIMD width of the device is 32, it's interesting that larger groups are favored. This is likely again related to memory locality. With regard to the tiled approach, I would expect the best performance when roughly all positions for all concurrently executing tiles fit into the card's cache. I would need more information than I can find about the card specs to check this hypothesis though. This may be another reason that performance usually drops for the larger group sizes.
Code
For anyone who cares, the source code is available.
I can only guarantee it will compile and run on linux with an NVIDIA OpenCL device because that's all I've tried it on.
Parallelism Competition Presentation
References
- Steve Plimpton, Fast Parallel Algorithms for Short-Range Molecular Dynamics, Journal of Computational Physics, Volume 117, Issue 1, 1 March 1995, Pages 1-19, ISSN 0021-9991, 10.1006/jcph.1995.1039. (http://www.sciencedirect.com/science/article/pii/S002199918571039X)
- Kevin J. Bowers, Edmond Chow, Huafeng Xu, Ron O. Dror, Michael P. Eastwood, Brent A. Gregersen, John L. Klepeis, Istvan Kolossvary, Mark A. Moraes, Federico D. Sacerdoti, John K. Salmon, Yibing Shan, and David E. Shaw. 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE conference on Supercomputing (SC '06). ACM, New York, NY, USA, , Article 84 . DOI=10.1145/1188455.1188544 http://sc06.supercomputing.org/schedule/pdf/pap259.pdf
- Adventures in OpenCL
- UCDavis ChemWiki
- NVIDIA GPU Gems 3, Chapter 31. Fast N-Body Simulation with CUDA
- UMD Physics notes
List of Work By Each Student
All work here is my own, with the exception of where code is noted as being borrowed from another source (this includes most of the OpenGL stuff and some utility functions).